22q11.2 Deletion Syndrome: The Most Common Microdeletion Syndrome
22q11.2 deletion syndrome, sometimes called DiGeorge syndrome or velocardiofacial syndrome, is a chromosomal condition that can lead to a wide range of developmental delays and ongoing health problems.1 Health problems caused by the syndrome can resemble a number of other medical conditions, so when not identified in the womb, children with 22q11.2 deletion syndrome often experience a long medical journey before receiving an accurate diagnosis. However, prenatal screening can lead to earlier detection during pregnancy. This can enable treatment in early infancy, which can reduce the severity of the condition and lead to healthier lives for affected children.
A recent study of over 18 thousand of pregnancies published in the American Journal of Obstetrics & Gynecology demonstrated that 22q11.2 deletion syndrome is more common than previously reported.2 Read on to learn more about 22q11.2 and how to identify it during pregnancy.
What are chromosomal conditions?
The genes in our DNA provide the instructions for how our bodies grow and function. Inside our cells, our DNA is packaged into tightly wrapped structures called chromosomes. Humans usually have 23 pairs of chromosomes. We inherit one set from each parent, for 46 chromosomes total.
A chromosomal condition is a type of genetic condition that occurs when there are extra or missing chromosomes (aneuploidy) or pieces of chromosomes (duplications or deletions). For example, trisomy 21 (Down syndrome) is caused by an extra chromosome 21. Chromosomal conditions are often associated with developmental delays, cognitive delays, or congenital anomalies (birth defects). In some cases the symptoms are mild, in others they require intensive medical support. Most chromosomal conditions occur by chance. They are caused by random errors in an egg or sperm, atypical events during fertilization, or abnormal cell division immediately following conception.
What is a microdeletion?
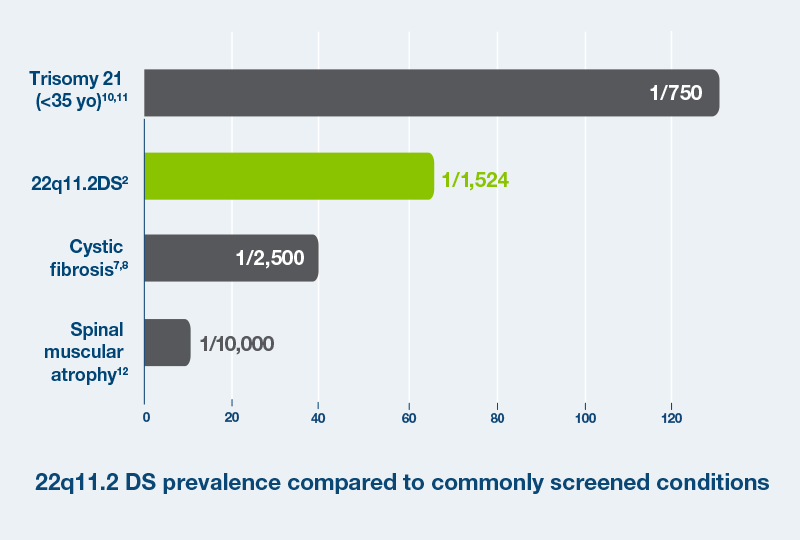
A deletion is a type of chromosomal condition in which a piece of a chromosome is missing. When the missing piece is too small to be seen under a microscope, it is called a microdeletion. Depending on what DNA is missing, syndromes caused by deletions and microdeletions can have a range of effects, from minor cognitive delays to severe ongoing organ health problems. Researchers have identified more than 200 microdeletion-associated syndromes, many of which are extremely rare.3 22q11.2 deletion syndrome is the most common of these, affecting approximately 1 in every 1,524 pregnancies, according to a recent study of over 18,000 pregnancies.2
What is 22q11.2 deletion syndrome?
22q11.2 deletion syndrome can involve many developmental, medical, and learning conditions, including:1
-
Heart defects
-
An underactive thyroid gland, which can cause delayed growth and impaired bone development or immune function and frequent infections
-
Autoimmune disorders
-
Cleft palate
-
Gastrointestinal problems and difficulty gaining weight
-
Breathing difficulties
-
Muscle weakness
-
Missing infant developmental milestones
-
Hearing impairment
-
Delayed speech
-
Learning difficulties
-
Behavioral and mental health difficulties
Until recently, 22q11.2 syndrome was thought to affect 1 in 3000 to 1 in 6000 infants.4,5 However, a recent study has shown that 22q11.2 is much more common, occurring in 1 in 1,524 pregnancies.2 This rate is comparable to other conditions commonly screened for during pregnancy, such as cystic fibrosis (1 in 2,500 pregnancies).6-8
What causes 22q11.2 deletion syndrome?
22q11.2 deletion syndrome is caused by a microdeletion in a specific region of chromosome 22. The size of the deletion varies and can result in up to 40 missing genes. The number and severity of symptoms depends on the location and size of the deletion. In most cases, 22q11.2 deletion syndrome is not inherited, meaning that in most cases neither parent of an affected child has the syndrome. However, people with 22q11.2 deletion syndrome can have children and pass the syndrome on to them.9 Most cases occur at random, caused by an error during the formation of a sperm or egg cell.
Since the condition mostly occurs at random, the baseline risk of 22q11.2 deletion occurring in a pregnancy is low. However, if either parent has the syndrome, the risk of passing it on to a child is 50%.
Can 22q11.2 deletion syndrome be treated?
While 22q11.2 deletion syndrome cannot be cured, early treatments can greatly improve a child’s health and quality of life.
Unlike many other genetic conditions, it is not always obvious that an infant has the condition at birth, and some treatments need to be performed before symptoms are apparent. In some cases, a baby will need urgent surgery at birth. Potential treatments facilitated by early identification include:1,2
-
Modified labor and delivery plans for a hospital birth with high-risk specialist support
-
Preplanned surgeries for heart defects
-
Treatment of low calcium levels beginning immediately at birth to prevent seizures and aid bone development
-
Surgical correction of palate abnormalities to help breathing, feeding, and speech
-
Customized care strategies for immune deficiencies, developmental delays, and learning difficulties
A tailored treatment plan starting at birth can make a big difference in a child’s growth and health. For example, one of the common symptoms of 22q11.2 deletion syndrome is hypocalcemia, or low levels of calcium. Low calcium levels can cause affected babies to have repeated seizures, likely due to calcium’s role in normal brain function. Seizures in young infants are often difficult to detect and likely to go untreated. Repeated seizures can damage growing neurons and are linked with an increased risk for intellectual impairment in adulthood. Early treatment with calcium can help reduce seizures and prevent long term neuronal damage. When 22q11.2 deletion syndrome has not been diagnosed early and seizures go undetected, it is common for providers to miss hypocalcemia.3
How is 22q11.2 deletion syndrome diagnosed?
The most common path to a 22q11.2 deletion syndrome diagnosis starts with a prenatal screening risk assessment. If screening shows a high risk of the syndrome, diagnostic testing should be performed to obtain a definite diagnosis.
If 22q11.2 deletion syndrome is diagnosed prenatally, your provider could recommend consulting with a high-risk pregnancy specialist, considering prenatal treatments, and updating your birthing plan to give your baby the best possible start.
If first detected after birth, diagnosis also requires genetic testing. A healthcare provider could recommend testing for infants born with severe heart defects and cleft palates. However, many children with 22q11.2 deletion syndrome are not diagnosed until they are toddlers with more pronounced developmental delays. Older children gain fewer benefits from targeted interventions like calcium supplementation. Prenatal testing provides the best path to early treatment and interventions for children with 22q11.2 deletion syndrome.
What is prenatal screening?
Prenatal screening tests help determine the risk of a chromosomal condition occurring during a pregnancy. Today, the most common type of screening is noninvasive prenatal testing, or NIPT. The American College of Obstetricians and Gynecologists currently recommends NIPT screening for all pregnancies for the most common chromosomal conditions.
When you are pregnant, DNA from your baby’s placenta passes into your bloodstream. NIPT uses a standard blood draw to detect and analyze this fetal DNA with no risk to the pregnancy. NIPT can be performed as early as nine weeks in pregnancy to help identify when a high-risk specialist should be involved.
SNPs are the 1% of DNA that make people different from one another. NIPT screens that use SNP-based technology, like PanoramaTM NIPT, can identify what DNA comes from the baby and therefore can identify signs of potential chromosomal conditions with high accuracy. By combining this information with other known risk factors, such as parental age and family history, NIPT can create a highly personalized risk profile for multiple chromosomal conditions.
Can prenatal screening with NIPT diagnose 22q11.2 deletion syndrome?
Prenatal screening, including NIPT, is not intended to diagnose any disease or condition. Instead, these tests are designed to help narrow down which pregnancies are most likely to benefit from additional diagnostic testing, like chorionic villus sampling (CVS) or amniocentesis. Many diagnostic tests require the support of a specialist and can slightly increase the risk of miscarriage. Screening helps reduce the need for diagnostic testing.
If your NIPT screen results indicate a high risk for 22q11.2, diagnostic testing can clarify whether the condition is present. You should speak with your healthcare provider to learn about different diagnostic testing options.
What are the benefits of identifying 22q11.2 deletion syndrome in the womb?
Early detection of 22q11.2 deletion syndrome can help parents and their babies’ healthcare team prepare to provide targeted care shortly after birth. Certain surgeries and treatments are most effective when performed immediately following birth. Early knowledge ensures that surgical preparations are made and important treatment windows are not missed.
Complex conditions like 22q11.2 deletion syndrome often require a multidisciplinary team of healthcare professionals to manage the range of potential problems associated with 22q11.2 deletion syndrome. An early diagnosis gives families time to identify specialists, connect with support groups, apply for government assistance, and adjust insurance coverage when possible.
How accurate is Panorama NIPT for detecting 22q11.2?
Natera’s Panorama NIPT uses SNP-based technology to analyze your baby’s DNA from cell-free DNA floating in your bloodstream, with zero risk to the health of your pregnancy. It is the only NIPT that can distinguish between a parent’s and baby’s DNA for more accurate results. For example, Panorama is the only NIPT that can reliably detect triploidy, a genetic condition caused by the presence of an extra full set of 23 chromosomes. Because of the high resolution of SNP-based screening, Panorama detects >99.9% of the most common type of 22q11.2 microdeletions (larger missing sections of DNA) and 83.3% of all 22q11.2 microdeletions (including smaller missing sections of DNA).2
Despite its high detection rate, Panorama is not a diagnostic test for 22q11.2 microdeletions. Panorama tells pregnant people the risk that their baby has 22q11.2 deletion syndrome. Although receiving an incorrect result is uncommon, it is possible: For 22q11.2, a false positive (reported as “high risk”) occurs in approximately 1 in every 2000 tests (0.05%).2 This rate of false positive tests means that a positive result for 22q11.2 deletion syndrome has a positive predictive value (PPV) of 53%. In other words, if a pregnancy receives a positive or “high risk” result, there is a 53% chance that the baby actually has the syndrome. Additional diagnostic testing is necessary to confirm if the condition is present.2
While rare, false negatives are also possible. In a study of 18,289 pregnancies, Panorama gave “low risk” results in 2 cases which were later diagnosed with 22q11.2 deletion syndrome.2 This rate of false negatives means that Panorama has a sensitivity of 83.3% for the syndrome, meaning there is an 83.3% chance that a negative or “low risk” result is accurate. The potential for false negatives is part of the reason that other forms of prenatal monitoring, such as regular checkups and ultrasounds, are essential for a healthy pregnancy.
Learn More
Natera is a leader in personalized genetic testing and diagnostics. As part of our commitment to our patients, we provide complimentary information sessions with board-certified genetic counselors. Our counselors are available to answer questions from 6 AM to 5 PM Pacific Time, Monday through Friday, excluding holidays. Please call 866-985-4336 and ask to speak with the on-call genetic counselor. You may also email gc@natera.com with questions.
References
1National Library of Medicine. 22q11.2 deletion syndrome. Genetics Home Reference. https://ghr.nlm.nih.gov/condition/22q112-deletion-syndrome#genes.
2Dar et al. Am J Obstet Gynecol. Epub prior to publication. https://doi.org/10.1016/j.ajog.2022.01.002
3Weise et al. J Histochem Cytochem. 2012;60(5):346-358. https://doi.org/10.1369/0022155412440001
4Botto et al. Pediatrics. 2003;112(1 Pt 1):101-7. https://doi.org/10.1542/peds.112.1.101
5Olsen et al. Lancet Psychiatry. 2018; 5(7):573-580. https://doi.org/10.1016/S2215-0366(18)30168-8
6Cheung et al.. Genet Med 2014;16, 40–44. https://doi.org/10.1038/gim.2013.71
7Hamosh et al. J Pediatr. 1998;132(2):255-259. https://doi.org/10.1016/S0022-3476(98)70441-X
8O’Sullivan. Lancet. 2009;373(9678):1891-1904. https://doi.org/10.1016/S0140-6736(09)60327-5
9Poirsier et al. Eur J Hum Genet. 2016 Jun; 24(6): 844–851. https://doi.org/10.1038/ejhg.2015.219
10UpToDate. Maternal age-related risk for common fetal trisomies across pregnancy. Accessed Jan 20 2022. https://www.uptodate.com/contents/image?imageKey=OBGYN%2F75423
11Centers for Disease Control and Prevention. National Vital Statistics Reports, Vol. 70, No. 2, March 23, 2021.
12Prior et al. Spinal muscular atrophy. In: Adam et al, eds GeneReviews. February 24, 2000. Updated December 3, 2020.